Breaking the Mold: Expanding the Horizons of Medicinal Chemistry


In the field of medicinal chemistry, change comes slowly. Every few years, someone with access to vast pharmaceutical data conducts a statistical analysis, aiming to identify new rules for drug-likeness. Yet, ironically, these rules seem to evolve more than the field itself. As a medicinal chemist, I've observed our tendency to dismiss clinically successful compounds that don't fit our rigid rulebooks. This dogma extends to creating separate rules for different modalities - one set for PROTACs, another for small molecules.
This article isn't about reviewing rules beyond Lipinski's Rule of Five or proposing new ones. Instead, it's a candid exploration of the untapped potential lying beyond these self-imposed boundaries. Deep down, most medicinal chemists recognize the promise of macrocycles, peptide analogues, and chimeric compounds like PROTACs. Yet, many shy away from these possibilities, constrained by industry dogma, and the daunting challenge of optimizing such molecules into drugs.
Our field is highly empirical and based on available data. However, unlike other scientific disciplines that regularly challenge empirical evidence with new experiments and hypotheses, medicinal chemistry often remains stubbornly set in its ways. A few outliers beyond Rule of Five (bRO5) or similar guidelines are easily dismissed by seasoned chemists. Even when a team of chemists takes on the challenging problem of going beyond the rules and work on challenging macrocyclic compounds, it takes years of synthetic effort to collect data on just a few thousand compounds. The hurdles don't end with synthesis. When we do create larger bRO5 compounds, our current assays often fall short. The emerging field of PROTACs has revealed how larger compounds can introduce new challenges in our routine pharmacokinetic screening cascade, yielding unreliable results. This begs the question: Are we merely operating in a comfort zone, much like someone searching for lost keys under a streetlamp because it's too dark to look elsewhere?
The Untapped Potential
Modern drug discovery with small molecules may have reached a local minimum. We’ve tried to target majority of known proteins with a binding pocket, from kinases to bromodomains to protein-protein interactions (PPIs) with buried surfaces. Medicinal chemistry had its golden era during the boom of kinase drug discovery. However, we now know that many blockbuster kinase drugs are not as selective as originally intended. The field was fortunate, as these unselective compounds turned out to be highly effective medicines. But new targets, emerging from the ever-evolving fields of genomics, epigenomics, transcriptomics, and proteomics, may not afford us the same luxury. The biology of diseases offers far more than kinases and targets with deep pockets – think of PPIs with larger interfaces, transcription factors, helicases, and beyond. Slightly larger compounds, such as macrocycles, peptides, and chimeric compounds hold the potential to create highly potent and selective drugs.
I’m encouraged to see targeted protein degradation becoming mainstream in the industry after initial scepticism from traditional medicinal chemists. If you had shown early PROTACs to a medicinal chemist, they were likely to have dismissed them as mere chemical biology tools. I must admit, I was once one of those sceptics, as I mentioned in my article on RNA-targeting small molecules, where I highlighted how different these RNA-targeting compounds are compared to known molecules with favourable pharmacokinetic properties.
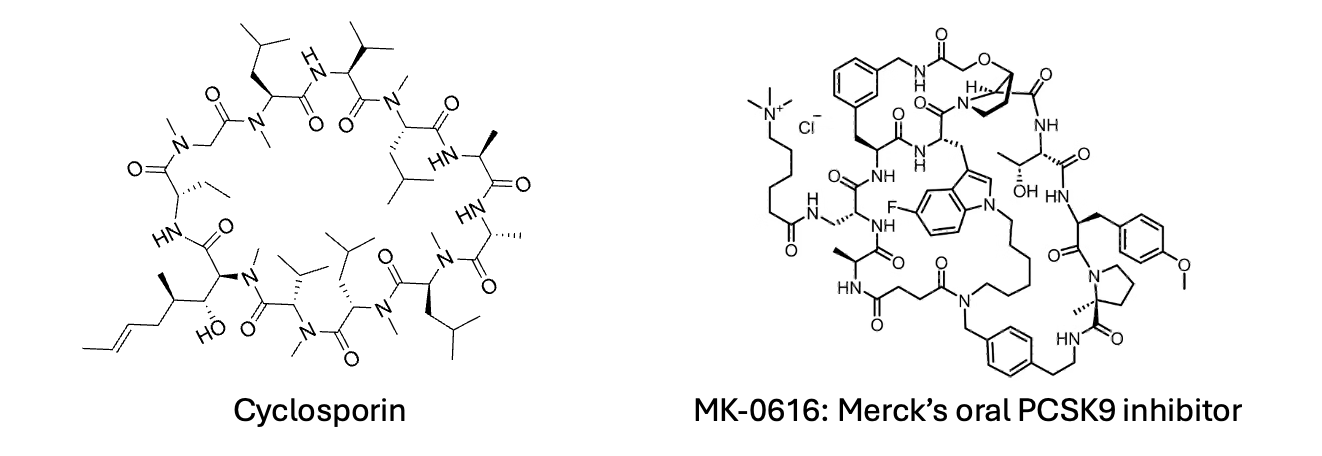
Many researchers have ventured into cyclic peptides and macrocycles, but progress has been slow and linear, a few hundred compounds are produced per lab each year. These larger compounds offer the advantage of being exquisitely potent and selective. Permeability is a significant challenge with these larger molecules, and they are notoriously difficult to optimize. However, there are examples of macrocyclic compounds that can cross membranes, often by hiding hydrogen bond donors through intramolecular hydrogen bonding or by behaving as chameleons, like cyclosporin, which can conceal its polar surface by changing conformation in lipophilic environments such as lipid bilayers when crossing biological membranes. But is it almost impossible to design such compounds de novo? Have we truly reached a global minimum in drug design with synthetic small molecules, with Cyclosporin and oral macrocyclic PCSK9 inhibitors being mere outliers? Or is it time for a concerted effort to explore what lies beyond our current boundaries?
The Optimization Conundrum
Optimizing even small synthetic molecules into viable drugs often requires a lengthy 3-5 years of dedicated synthetic chemistry, with teams of chemists meticulously synthesizing thousands of compounds. Consider this: a single molecule typically presents 5-10 or more pivot points, each with 10-20 potential variations. The resulting combinatorial explosion creates a universe of possibilities far beyond what any chemist could hope to synthesise in a lifetime.
Yet, we persist in this linear approach, year after year, barely scratching the surface of the chemical universe. Hand the same challenge to two distinct teams, and they'll inevitably traverse different paths, arriving at divergent outcomes. Our methodology isn't just inefficient – it is fundamentally limited in its ability to harness the full spectrum of possibilities. This isn't merely a challenge; it's a clarion call for transformative thinking.
Pushing Boundaries
We know the medicinal chemistry of small molecules as practiced in the industry works and it has yielded many blockbuster medicines. But should we ignore any other targets that demand much larger molecules, or necessitate solutions involving other modalities? Should we ignore the Cyclosporins of the world? My answer is a resounding 'No.' We should do much more, but it's easier said than done. The optimization problem of macrocycles or larger compounds like cyclic peptides is enormous compared to small molecules of around 500 Daltons molecular weight. Cyclosporin, the poster child for beyond-rule-of-five drugs, is a natural product resulting from millions of years of evolution. One might argue that replicating evolution is not worth the effort, but we have seen successes in the field of directed evolution. Methods like phage display and mRNA display are effective in identifying very potent binders to target biomolecules. I was fortunate enough to work on such a project led by my former colleague Kim Liu, now a synthetic biologist at MRC-LMB. The challenge is that molecules derived from these display technologies often suffer from poor pharmacokinetic properties, such as metabolic stability and permeability. However, the advantage of these larger molecules is their generally high potency and selectivity for their targets. The unique conformations of large macrocycles mean there is a reduced chance that other targets will fit the same constrained structure. Optimizing these compounds is extremely challenging due to their size and the vast number of possible modifications. Alanine scans, and reliance on crystal structure can sometimes lead you to an optimised compounds quickly but often its very challenging. For example, in a simple 10-mer single-cycle macrocycle, you can make changes at every position with non-classical amino acids or different chemistries other than amide couplings to connect each monomer, resulting in millions of possibilities. So, is it even feasible?
The Path Forward: Innovative Solutions and Persistent Challenges
While engineering biological systems to perform reactions beyond amide coupling is an exciting prospect, the reality is more complex. Current mRNA display technologies, which are gaining traction, especially among teams focused on peptides, do offer some unnatural amino acids, but the chemistry remains largely limited to amide couplings. Jason Chin's lab at the LMB is doing groundbreaking work in this area, pushing the boundaries of what's possible with tRNA and ribosome-like systems. They are even envisioning harnessing unnatural chemistries beyond amide coupling within living cells by expanding the genetic code. However, this technology is still in its early stages and not yet ready for practical drug discovery campaigns. Nonetheless, Jason Chin’s recent founding of Constructive.bio suggests that significant advancements could be on the horizon, so this is definitely a space to watch.
Even if we perfect these display technologies, a significant challenge remains: intracellular targets. How do we find compounds that can both enter the cell and bind to the target? Most current display technology screenings are conducted in buffers or simulated media outside of cells. This approach might yield a tight binder to your target, but it could take millions of compounds to find one that is potent, stable and capable of crossing the cell membrane.
Consider the oral PCSK9 inhibitor from Merck, often cited as a success story of macrocycles. Even this "poster child" targets an extracellular protein - getting these larger molecules into cells remains a formidable challenge. While techniques exist to enhance peptide permeability, such as adding positively charged amino acids, the story of enhancing peptide permeability is complex and is a topic for another day. Our focus here is on discovering larger molecules that can be made stable and permeable without resorting to large modifications with various tags.
Technologies like SICLOPPS (Split-Intein Circular Ligation of Peptides and Proteins) offer an intriguing approach, synthesizing cyclic peptides inside cells. However, the chemistry is currently limited to natural amino acids. In theory, you could generate an intracellular peptide and use a reporter assay to identify hits. But here's the catch: synthesis inside a cell doesn't guarantee cell permeability. Most proteins that function perfectly well inside cells cannot cross the cell membrane from the outside.
Some computational approaches have shown promise in designing macrocycles with favourable properties. David Baker’s lab has made notable progress in this area. However, balancing biological activity with favourable pharmacokinetic properties remains a significant challenge for these models. Designing macrocycles or cyclic peptides that fit within a binding pocket while also maintaining desirable pharmacokinetics is no easy task.
DNA-Encoded Libraries: A Promising Frontier
DNA-encoded library (DEL) screening could be a promising way forward. By synthesizing diverse DELs that include larger compounds, such as macrocycles and cyclic peptides with unnatural chemistries beyond simple amide couplings, we could potentially overcome many of the current limitations in the field. DEL offers an advantage over current display methods, as it allows for a broader range of chemistries. DELs have their own challenges, particularly in synthesizing longer oligomers, as the overall yield can be very low. However, innovative work coming out of Zurich, such as PureDEL, utilizes advanced chemistries with multiple linkers to purify DELs, allowing for the creation of larger peptides and macrocycles with higher purity.
In principle, you could grow cells in microfluidic drops and incubate them with on-bead cleavable DEL libraries. This would require a significant amount of engineering to make all the systems work together seamlessly. But almost every piece of the puzzle, from on-bead cleavable DELs to growing cells in microfluidic drops, has been solved separately. The challenge lies in integrating these solutions. Issues like material leakage from one bead to another, perfect cleavage of DEL members, and cell compatibility with microfluidic drops must be addressed. Despite these challenges, this method holds promise for finding cell-penetrant, on-target macrocycles and cyclic peptides in a single step. For those interested, I highly recommend looking into the work from Brian Paegel’s lab.
Decades of medicinal chemistry knowledge are invaluable in designing these libraries. On-demand DELs can be prepared based on initial hits, allowing iterative screens to find the best possible leads, mimicking evolutionary selection. Medicinal chemists can significantly impact the design of such DEL libraries by focusing on properties of individual monomers, intramolecular hydrogen bonds, cyclization chemistries to restrict conformation, and more. Even with these techniques, further optimization for other pharmacokinetic (PK) properties may be necessary, as cell penetration alone will not be sufficient.
Screening decks in many companies are typically filtered to exclude compounds that don't follow internal drug-like rules. This approach makes it unlikely to identify the next cyclosporin or Merck’s PCSK9 inhibitor. To boldly tackle novel biological targets without classical deep binding pockets, we need a combination of hit finding using synthetic macrocycles via DELs and/or unnatural amino acids through display technologies such as mRNA display. While several companies have made some efforts in these areas, many campaigns stall at the initial hit identification phase due to the complexities in improving PK properties of large compounds.
Given the millions of ways to modify a large macrocycle or cyclic peptide, traditional synthetic efforts, producing ~1000 compounds a year, may not be sufficient. On-demand DEL screening could revolutionize this process, allowing for optimization on a scale of millions rather than thousands. Combining this with machine learning on the resulting data could significantly speed up hit identification and optimization.
This is a challenging problem that deserves more funding, long term vision, and attention. For venture capitalists willing to take on tough, high-risk projects, an investment of $3-5 million in seed funding could be the catalyst for groundbreaking advancements. While the challenges are significant, the potential rewards in terms of novel drug discoveries could be enormous.
As we stand at this crossroads in medicinal chemistry, it's clear that our traditional approaches, while successful, may be limiting our potential. The challenges are immense, but so are the possibilities. By embracing new technologies like microfluidics, DEL screening, and other display technologies, expanding our chemical space, and challenging our long-held assumptions, we can usher in a new era of medicinal chemistry.
I would like to thank Kim Liu for his invaluable insights and thoughtful feedback!