Generative Chemistry for Everyone: A Hands-On Guide using SAFE Encodings


Generative AI has made remarkable strides in recent years, revolutionising fields like image, text, and even video generation. These advancements are largely due to the vast amounts of data available for training these models. However, in the realm of generative chemistry, there isn't the same volume of data to learn molecular representations of drugs or drug-like molecules. As a result, generative chemistry, in its current state, cannot yet match the expertise of a human medicinal chemist. However, recent advancements have propelled generative chemistry tools into a valuable position within drug designers’ toolbox. In this article, I aim to introduce the generative chemistry pipeline with a specific example of linking two fragments to generate a novel scaffold.
Here is a link to the Google Colab notebook that you can use to generate novel molecules, no coding experience required. I have added comments in the Colab notebook to make it accessible to everyone. Whether you are a student, a fellow drug discovery scientist, or a curious mind eager to dive into generative chemistry through a hands-on example, you’ve come to the right place. For those who have never used Python or Notebooks before, don't worry, I’ve got you covered! Check out the tips at the end of this article to help you get started.
Generative chemistry uses generative AI to produce molecules with desired properties. Imagine DALL-E, but for molecules. That's generative chemistry in a nutshell. These tools for de novo drug design have been capturing headlines, often touted as the next big thing that might replace human chemists. This sensationalism has led to mixed reactions, with some scientists becoming sceptical and dismissing these tools as mere hype. While it's true that replacing human chemists with current technology is still a distant dream, I believe these tools can significantly enhance drug discovery programs when used correctly.
The most commonly used open-source generative chemistry tools typically follow these steps:
· AI models such as RNNs and transformers learn to "speak" molecule, using languages like SMILES or graphs. In this article, we’ll focus on a specific language representing molecules called Sequential Attachment-based Fragment Embedding (SAFE).
· Reinforcement learning then plays the role of a molecular architect by guiding the generation of molecules towards desired objectives.
Current generative chemistry tools face two major hurdles:
· Synthetic feasibility: While scores exist to measure synthesizability, they're far from perfect. Fortunately, this is an active area of research, promising future improvements.
· The Reinforcement Learning (RL) conundrum: Crafting an effective scoring function for RL is like trying to explain "good taste" to an AI. The algorithm often latches onto quirks that boost scores but produce molecules that leave medicinal chemists scratching their heads. This creates a trust gap between chemists and these tools. For instance, using docking scores can lead to oversized molecules with excessive hydrogen bonds. Balancing this requires a delicate dance of additional parameters, like molecular weight and hydrogen bond counts. It's a time-consuming process of trial and error.
There are also diffusion-based methods that can generate molecules within the binding pocket. However, their successful application in live projects has yet to be demonstrated. For those interested in these methods, I highly recommend Patrick Walters' post, which highlights some of their shortcomings.
A hands-on guide using SAFE Encodings
Now, let's dive into a practical example to introduce you to generative chemistry through SAFE encodings. These encodings, introduced by Valence Labs, look very similar to SMILES but connect fragments with dots. This separation by dots makes SAFE particularly useful for common medicinal chemistry tasks such as scaffold decoration, scaffold hopping, fragment expansion, fragment merging, and linker design for chimeric compounds like PROTACs. You can read more about SAFE encodings in this paper and find detailed information in the documentation page.
Unfortunately, the open-source Python package for SAFE does not include scoring and reinforcement learning algorithms yet. We hope to see these features in future iterations. For now, we'll use what is available in the public repository.
Defining a Hypothetical Task
Let’s define a hypothetical task that a medicinal chemist might face during a drug discovery campaign. For this example, we'll reference a recent paper from AstraZeneca in the Journal of Medicinal Chemistry. In this paper, they identified compound 31 as a Cbl-b inhibitor and solved its crystal structure bound to Cbl-b. Our task will be to find a new scaffold for compound 31. Typically, you might seek a new scaffold for several reasons:
· To improve pharmacokinetic (PK) properties of compounds if the current scaffold causes poor PK.
· To develop a backup series.
· To link two fragments occupying different parts of a binding pocket in the target protein.
· To explore novel chemical space during early hit identification stages, expanding on hits identified in a screening campaign.
Steps to Identify a New Scaffold
We will follow these steps to identify a new scaffold for compound 31:
- Import the 3D structure of compound 31 bound to the Cbl-b protein (PDB: 8qtk). The SDF file for the 3D structure of compound 31 is provided.
- Generate molecules with alternative scaffolds using SAFE from Valence Labs.
- Apply physicochemical and substructure filters to the generated compounds.
- Identify molecules that match the shape and electrostatics of compound 31 in its bound conformation from the crystal structure (PDB: 8qtk).
- Overlay and visualise the top-ranking compounds.
Follow along step by step using the Colab notebook. Your end results might vary from mine.
Firstly, let's upload the 3D structure of compound 31 from PDB: 8qtk. I have provided this SDF file, but you can do this programmatically using Python code or by using any of your favourite PDB visualisation tools. You can even try using your own ligand at this stage. If you do, you'll need to change the linking fragments as well, but I'll notify you when we get to that point. Compound 31 has a 3D shape with a central scaffold where a pyridone is bonded to a phenyl ring, acting as linkers to the fragments at both ends. The carbonyl in the pyridone interacts with the backbone of an amino acid in the protein. Our goal is to find a scaffold that provides similar vectors to connect the fragments at the ends while maintaining the overall 3D shape and electrostatics.
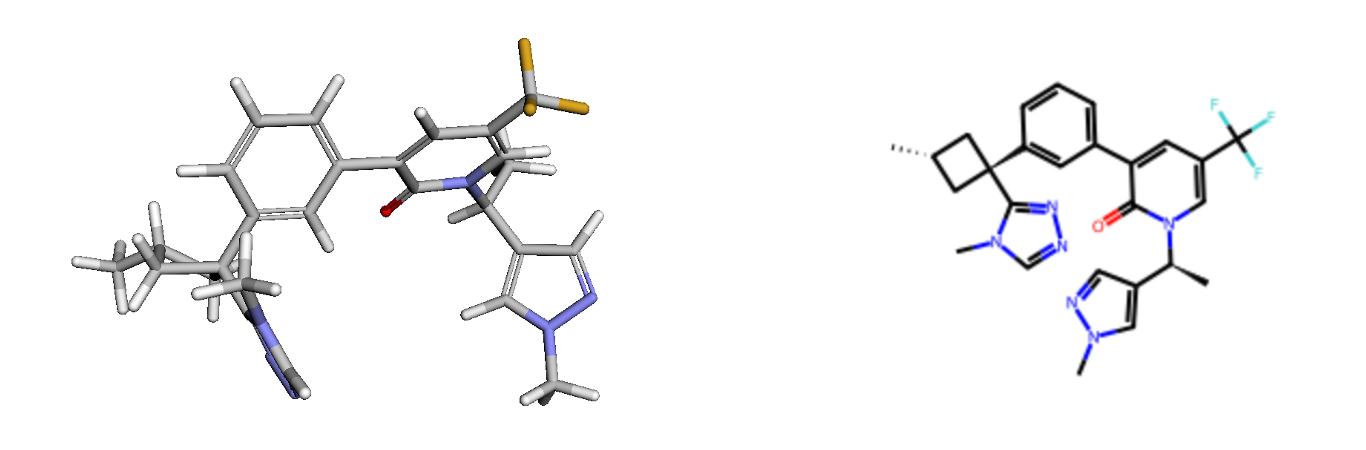
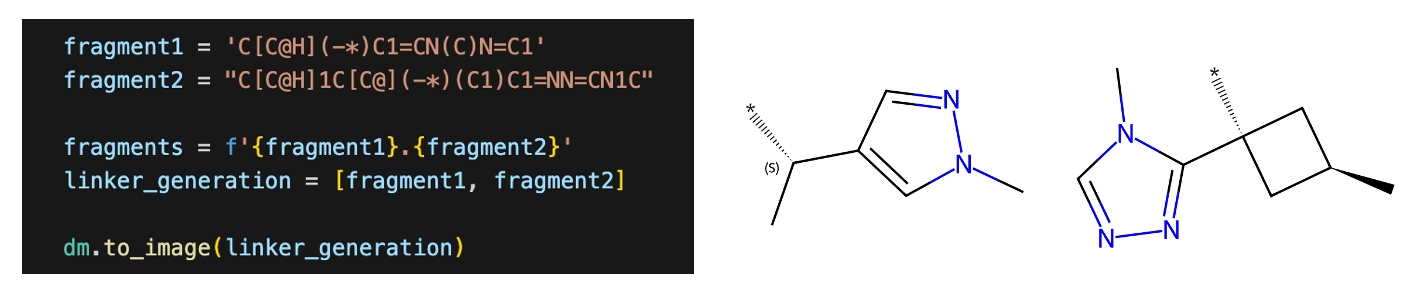
Next, the SMILES of fragments to connect the scaffolds are provided by removing the central scaffold and adding ‘*’ at the attachment points. You can add your own fragments at this stage if you decide to work on your own project rather than a hypothetical task here. SAFE’s two different methods, scaffold morphing and linker generation, are used to generate 1,000 compounds from each method, then the generated SMILES are combined.
I have also inserted the SMILES of the original compound 31 in the generated compound list as a control. Ideally, it should rank as the top hit after we complete our analysis, as a compound should have the highest 3D similarity with itself. While a perfect score of 1 might be expected for both 3D shape and electrostatic similarities, in practice, reproducing the exact conformation is challenging. Thus, we anticipate a score close to, but not exactly, 1.

Next, I remove duplicates, which eliminates more than half of the compounds. After that, physicochemical filters as well as ‘Dundee’ substructure filters are applied using Patrick Walters' useful_rdkit_utils package. Interestingly, most of the compounds pass the Dundee filters, which is already better than many other open-source generative chemistry alternatives.
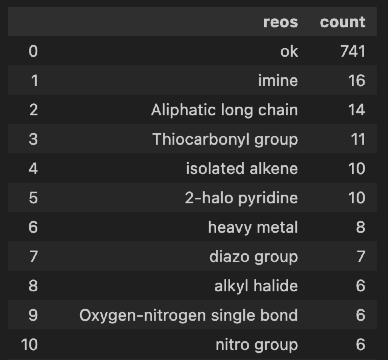
Then, I examine the rings found in the generated compounds again using Patrick Walters' useful_rdkit_utils package. I was pleasantly surprised to see that most of these ring systems look quite promising (ignore the first 3 rings as they are present in our fragments). With my synthetic medicinal chemistry background, I can tell, even without using complicated scores, that these rings are much better than those produced by several other open-source methods I have seen. Of course, you will find some odd ones if you scroll down the list, but in general, the list looks good to my synthetic chemistry eye.
Finding Compounds that Match the Shape and Electrostatics
Now, let's find compounds that match the shape and electrostatics of the original compound 31 as it is bound in the crystal structure. Using RDKit's conformer generation and the espsim package, I align 70 conformers for each compound with the 3D structure of compound 31 from the crystal structure. I then calculate shape and electrostatic similarities and combine them into a total similarity score. There are a few caveats to this method:
- Sampling Conformers: Even with 70 conformers, we might miss a low-energy conformer that could have a similar shape to compound 31. Ideally, you would want to sample a larger number of conformers or use better conformer ranking approaches. One package I highly recommend is Auto3D, but it can be slow for this implementation. If you have computational resources and time, Auto3D is excellent for generating conformers with energy differences between them.
- Conformer Energy: Even if the conformer of a generated compound aligns well with compound 31, it doesn't guarantee that the generated compound is in a low-energy conformer. You can use open-source tools like Auto3D to find low-energy conformers.
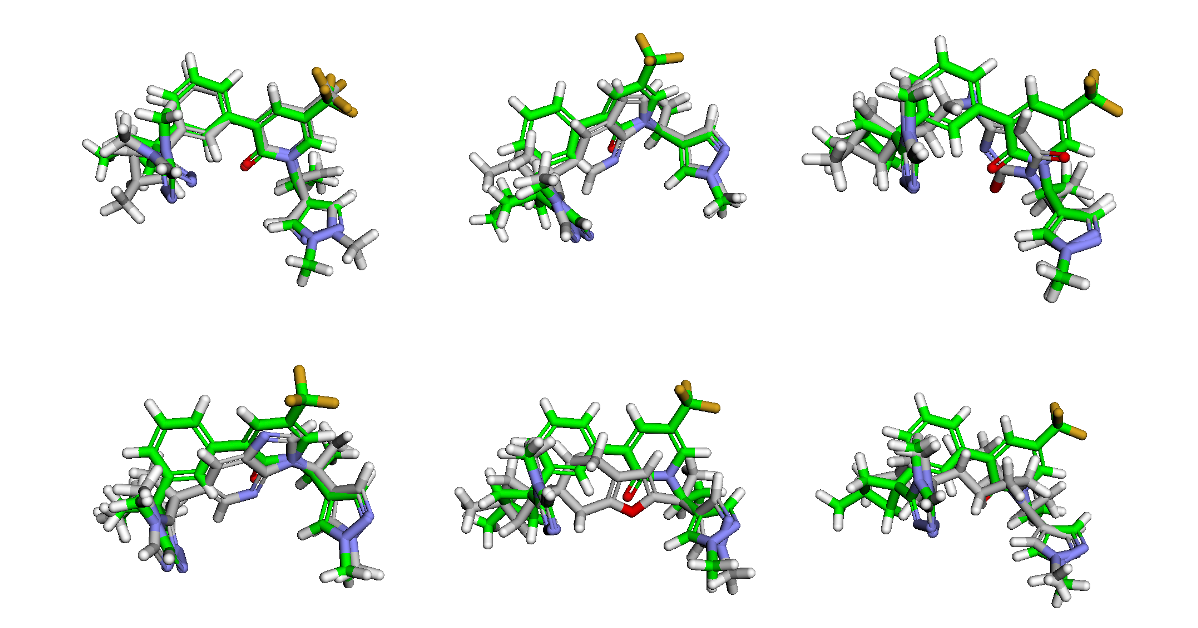
Despite these caveats, our method has been effective in identifying the top hit. Recall that we included the SMILES of compound 31 in our generated SMILES list as a test. The compound has survived the filters and is the top-scoring hit, as expected. Here are the top 6 hits with overlays on top of the original compound 31 (green).
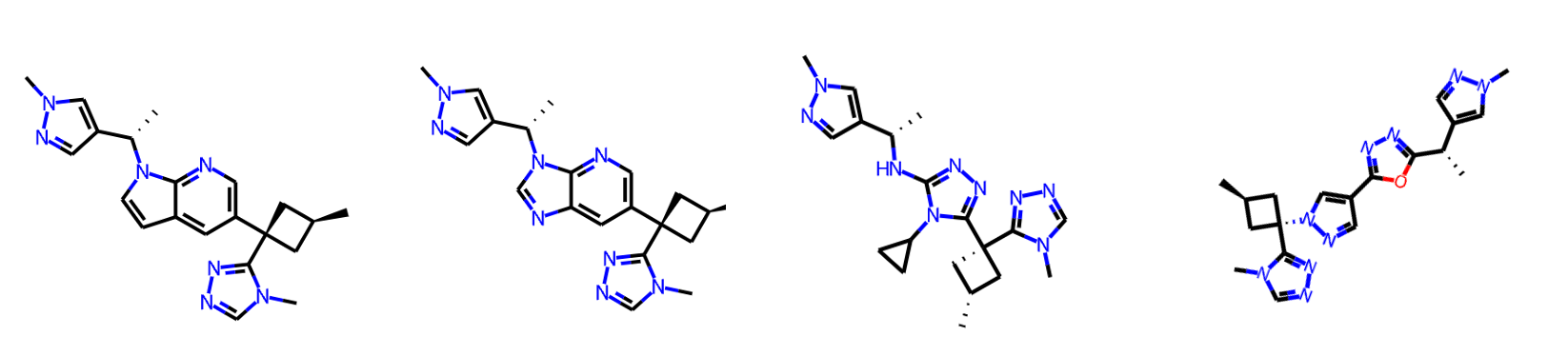
After quickly reviewing the top 20 hits, here are some of my favourites. With some tweaks to these scaffolds, such as adding the extra CF3 group that compound 31 has, some of these might work well. And, you can rerun the workflow to generate more ideas and combine the ones you like at the end.
Final Thoughts
To conclude, this was just a glimpse into the world of generative chemistry. As you may have noticed here or experienced in your projects, the intuition of medicinal chemists is incredibly valuable in identifying the right compounds. This is why I am particularly excited about research that incorporates humans in the loop. A notable paper from AstraZeneca colleagues explores this concept. In principle, it should be possible to have humans in the loop to rank the generated compounds and then refine the AI model accordingly. However, successfully implementing this approach requires many human medicinal chemists to invest significant time in reviewing generated ideas. This is very similar to how the field of text generation, with notable examples such as ChatGPT or Claude, have progressed by involving humans in the loop on a large scale.
How to Run a Google Colab Notebook
If you are completely new to coding and notebooks, don’t worry! Here’s how to open and run a Google Colab notebook:
Opening the Notebook
- Click on the provided link to open the Colab notebook in your browser.
- Sign in with your Google account if prompted. You will need to save the notebook in your Google drive if you want to make changes in the notebook and save them.
Running Code in the Notebook
Google Colab notebooks contain cells. Here’s how to run them:
- Run All Cells:
Click Runtime > Run all to execute all cells in the notebook. Do this if you don’t want to change anything in the code, such as adding your own sdf file or fragments. If you need to make changes, run individual cells as explained below.
- Run Individual Cells (recommended, so that you understand each step):
Scroll to the code cell (grey background with [ ] on the left).
Click the Play button or press Shift + Enter.