The Promises and Perils of Molecular Glues: Unpacking the Concept and Field’s Challenges

Molecular glues are making headlines, with millions of dollars pouring into deals that promise groundbreaking advancements in drug discovery. Companies like VantAI, Neomorph, and Monte Rosa are leading the charge, securing major partnerships with large pharmaceutical firms. But what exactly are molecular glues, how are they discovered, and why are they attracting so much attention now? Let’s dive in and explore.
What Are Molecular Glues?
Simply put, molecular glues are small molecules that "glue" or stick two proteins together. Molecular glue degraders (MGDs) are a class of molecular glues that stick a target protein to an E3 ligase complex, leading to the protein’s ubiquitination and subsequent degradation by the proteasome. This mechanism is akin to that of the more well-known proteolysis-targeting chimeras (PROTACs), which are bifunctional molecules with a linker that connect the protein of interest to the E3 ligase complex. However, molecular glue degraders are single molecules without any linker, which makes them distinct, though the difference is subtle, and not everyone in the field sees them as separate classes. While I won’t delve into the specifics of these definitions here, I plan to cover bifunctional PROTACs in a future article.
For those interested in the historical background of molecular glues, I highly recommend Stuart Schreiber's excellent review. He explores the history of the field, including the discovery of early glues with immunosuppressive properties like cyclosporin and FK506.
Small molecule drug discovery has primarily focused on targeting a single biomolecule, usually a protein with a deep binding pocket. This approach has been effective, but the landscape is changing. Advances in genomics, epigenomics, transcriptomics, and proteomics have revealed many disease-causing biomolecules that lack deep binding pockets, making them difficult to target with conventional small molecules. In a previous article, I discussed the potential of using unnatural cyclic peptides and macrocycles to target these challenging targets. However, optimizing these larger compounds to achieve the desired pharmacodynamic and pharmacokinetic profiles is difficult. This is where molecular glues come in, they offer a promising solution to targeting these "undruggable" proteins.
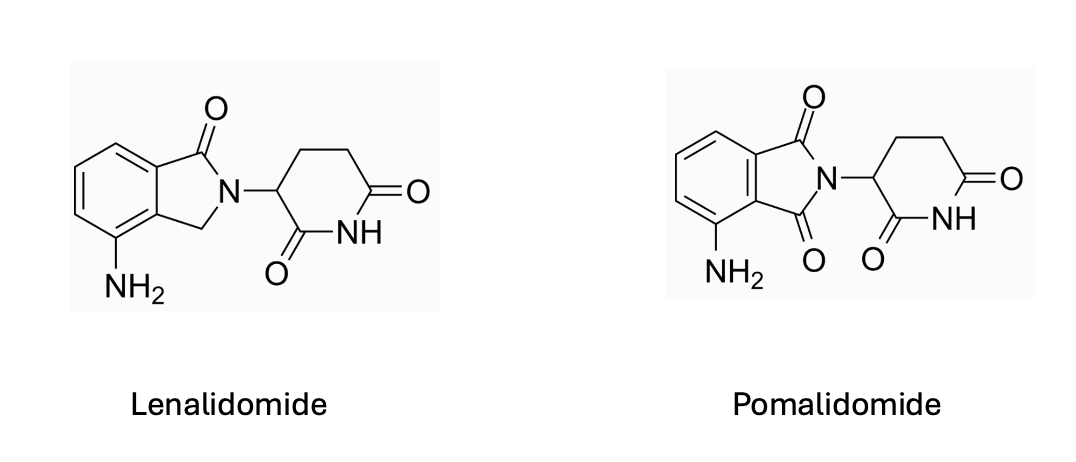
You might wonder how molecular glues, which are small molecules themselves, can target proteins that lack deep binding pockets. This becomes clearer when you consider lenalidomide (Revlimid), a thalidomide derivative that effectively treats multiple myeloma and several blood cancers. Lenalidomide induces the degradation of IKZF1 and IKZF3, transcription factors crucial in multiple myeloma. These transcription factors, like many others, lack deep binding pockets and are challenging to target with small molecules. However, lenalidomide manages to degrade them by "gluing" them to a component of the E3 ligase complex known as Cereblon (CRBN). Once attached, the E3 ligase complex tags these transcription factors for degradation by the proteasome.
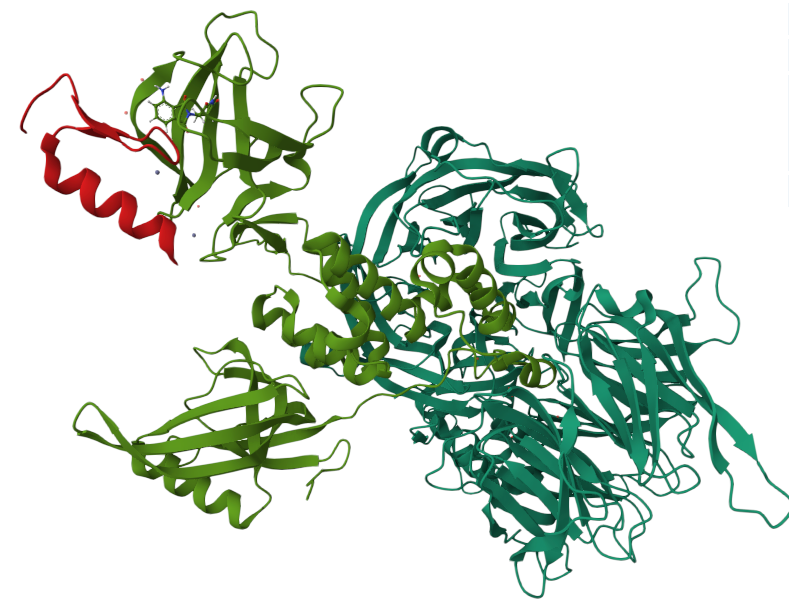
For those curious about the structural specifics of how the glue binds to IKZF1 and CRBN, this crystal structure of pomalidomide, a thalidomide analogue, provides valuable insight, particularly in showing the absence of a deep binding pocket in IKZF1 (in red in the image above) near the binding interface. It was later discovered that the degron, a unique sequence of amino acids recognized by E3 ligases, mimics a cyclic imide region of thalidomide analogues through post-translational modification of glutamine or asparagine residues. Check out this excellent paper from Christina Woo’s lab on the identification of CRBN’s degron.
The Quest for New Molecular Glue Targets
With the degron for CRBN identified, researchers can now computationally search for other proteins with similar degrons. For instance, in this paper from the Thomä lab, they have attempted to identify proteins that could be glued to CRBN. But can we extend this success to other E3 ligases? Can we computationally discover protein pairs that can be glued without detailed knowledge of their degrons? This is where things get tricky.
Many companies are attempting to map protein-protein interactions using various datasets, hoping to find potential glue-able pairs in these datasets. While this approach seems logical, it might not fully capture interactions that could be glued by small molecules. Some interactions are too weak to be detected without molecular glues, and may go unnoticed by current experimental techniques.
One common experiment involves pulling down proteins from a cell lysate that bind to a resin-bound E3 ligase. However, this method has significant limitations. First, not all potential glue-able proteins can be pulled down due to their weak affinity for the E3 ligase in the absence of a molecular glue. Second, and more critically, some proteins may not naturally bind to E3 ligases without specific post-translational modifications. Protein degradation in cells is tightly regulated, often through modifications like those observed for CRBN targets, as well as others such as phosphorylation, methylation, or acetylation. For these reasons, I’m somewhat sceptical of the hype surrounding AI-based companies that aim to identify these partners without thorough experimentation. I’ve tested some cutting-edge open-source AI-based multimer predictions, and even with known protein partners, these methods struggle. In some cases, degrons are located in the unstructured terminal regions of proteins. It’s hard to imagine how these "spaghetti" regions can be identified without knowing the exact degron sequence. Finding these protein pairs that can be glued is indeed a tough challenge, but it’s a challenge worth pursuing. Careful experimentation will be crucial in identifying these glue partners.
The challenges of Computational Approaches
Several companies have raised millions of dollars with the promise of using AI-based tools, like AlphaFold-Multimer, to identify protein partners that could be glued together. However, anyone who has tried using these algorithms for predicting protein-protein interactions will tell you about the challenges involved. While the idea of computationally finding protein pairs that can be glued by small molecules is exciting, it's far from simple.
The main issue is that these protein partners might not naturally bind to each other. They may require specific post-translational modifications to enable binding, which makes modelling them difficult. These modifications can take many forms, acetylation, phosphorylation, methylation, or even unique processes like the cyclization of side chains seen in CRBN targets, and they can occur at any of the numerous locations on various amino acids in a protein. While computational approaches are worth pursuing, it’s important to recognize that this task is anything but straightforward.
I've also heard discussions in various forums about using surface charges to identify potential protein pairs. While this approach sounds appealing, it could result in an overwhelming number of matches. Plus, post-translational modifications can alter surface charge density, complicating the results even further.
There’s a wealth of protein interaction data available in the public domain. Combining this data with information on degrons could indeed be useful in identifying potential protein partners for gluing.
But let’s say you’ve developed a brilliant computational pipeline that integrates all this publicly available data and ranks protein-protein interactions that could be glued, how do you validate these novel pairs experimentally? If your biophysical experiments, such as SPR or ITC, show that the pairs don’t bind with high affinity, would you discard them? Validating these pairs experimentally is no easy task, especially if the interaction is weak in the absence of a small molecule glue.
Is phenotypic screening the answer?
As we've explored, rationally designing experiments to find protein pairs that can be glued together is no easy feat. This challenge raises the question: could phenotypic screening be the key to identifying compounds that degrade your target protein? Interestingly, most molecular glues discovered to date have been identified through phenotypic assays that measure efficacy in carefully designed biological experiments. The mechanism behind these glues is often understood later, as was the case with thalidomide analogues and early glues like cyclosporin.
However, results from phenotypic experiments can be difficult to interpret. It's crucial to have well-thought-out control experiments to ensure that degradation is due to the primary effect of binding to the target protein, rather than due to secondary effects like reduced transcription of the gene or altered post-translational modifications. A recent paper from Novartis, led by James Bradner, showcases a successful phenotypic screening approach to identify a molecular glue degrader (MGD) for the WIZ transcription factor. In this study, they limited the screening to a library of thalidomide-like compounds to ensure that the mechanism of degradation was through gluing WIZ to CRBN.
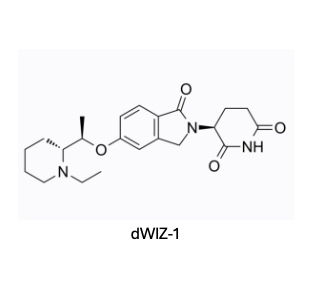
The team conducted meticulous control experiments to confirm the mechanism. For example, the compound dWIZ-1 showed no effect in erythroblasts where CRBN was knocked out via CRISPR-Cas9, indicating CRBN-mediated degradation. They also checked WIZ mRNA levels to ensure the reduction in protein was not due to decreased transcription. Additionally, they used innovative cellular experiments, like the NanoBit reporter system, to demonstrate that dWIZ-1 brings WIZ and CRBN together. Notably, the study found that WIZ did not bind to the CRBN complex at concentrations up to 10 µM without the glue dWIZ-1, reinforcing the idea that traditional validation methods like SPR might overlook valid targets. This paper is a must-read for anyone building a CRBN-focused library, and with many companies currently pursuing this direction, it's a timely resource.
A Thought Experiment: Discovering New Protein Pairs
Imagine this: to uncover protein pairs that don’t naturally stick together without molecular glues, what if we introduced a soup of diverse small molecules into the mix in pull-down experiments? Suppose you want to identify proteins that can be glued to a specific E3 ligase of interest. Why not create a diverse set of small molecules, perhaps thousands, and use them in the pull-down experiments?
Here’s how it could work: Immobilize your favourite E3 ligase and add a solution of 50-100 small molecules, each at a reasonable concentration of around 1 µM. Then, pull down all the proteins from the cell lysate of your choice and identify them using mass spectrometry. By conducting multiple such proteomics experiments, each with a different set of small molecules, you maximize your chances of finding protein partners that can be glued to the E3 ligase. While this experiment wouldn’t identify the specific small molecule responsible for gluing the proteins together, a separate screening process could be conducted later to pinpoint the exact glue for the targets identified in the proteomics experiment.
Many companies have now built their own libraries of small molecules targeting CRBN, and you can even purchase such libraries from commercial suppliers, such as this one from Enamine and this one from Mcule. Large-scale proteomics using these libraries, as described earlier, could be a powerful tool for identifying novel targets. However, it’s possible that we’ve already identified the only degron motif for CRBN, in which case we can computationally predict all proteins with this motif.
For less-studied E3 ligases, this type of proteomics experiment could be invaluable in discovering new targets that can be glued to the ligase. That said, it’s important to note that we have seen very few new molecular glues involving E3 ligases beyond the well-known ones such as CRBN, VHL, and DCAFs. Identifying molecular glues that work with other E3 ligases is clearly not trivial.
I’ve also come across few chemical biology approaches that involve artificially creating covalent bonds between protein partners, but that’s a discussion for another day.
Beyond Degradation: The Broader Potential of Molecular Glues
Let’s also consider the broader potential of molecular glues beyond protein degradation. Theoretically, molecular glues should be able to link two proteins that don’t naturally bind with high affinity. Instead of inducing degradation, these glues could alter the biology of the target protein by modifying its post-translational status, activating or inactivating it, or even changing its location within the cell.
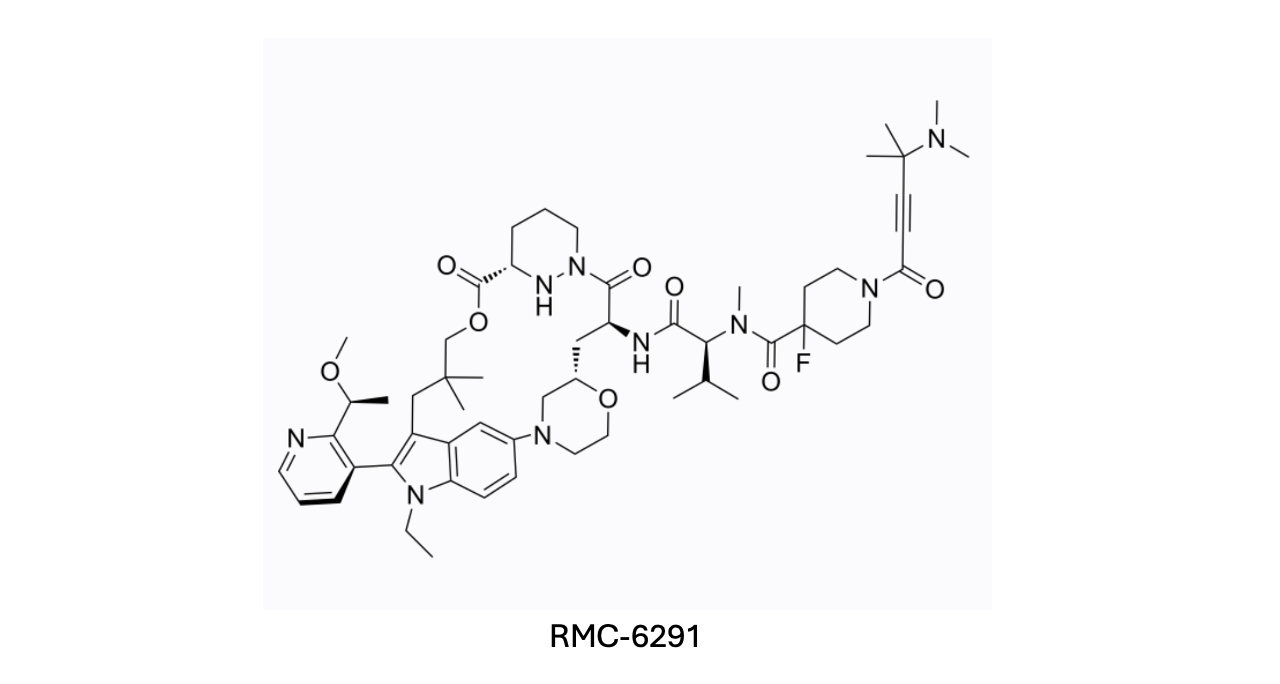
An exciting example comes from Revolution Medicines, where they identified a natural product Sanglifehrin A inspired macrocycle (RMC-6291) that glues KRASG12C (a common oncogenic mutation of KRAS) to cyclophilin A. This gluing inactivated the KRAS-mediated oncogenic signalling, leading to tumour regression in various human cancers. This discovery highlights the novel mechanisms molecular glues can unlock, proving that their potential is not limited to protein degradation.
To summarise, molecular glues offer a transformative opportunity to target proteins once considered ‘undruggable.’ With the surge in funding and interest in this field, we are likely on the brink of finding more rational methods to discover new targets that can be glued, potentially moving beyond the reliance on phenotypic approaches. The future of molecular glues is bright, and their applications may extend far beyond what we currently imagine.
This article has primarily focused on identifying protein pairs that can be glued together. In future discussions, I will delve into the screening methods for molecular glues, including DEL, ASMS, and other high-throughput techniques, once these protein pairs have been identified.